Quantification of biofilm and planktonic communities
Biofilms are difficult to quantify especially when microscopic observations are impractical. However it is important to quantify attached cell numbers to complement planktonic cell counts in order to achieve a holistic understanding of microbial processes. Traditional counting methods, including microscopic techniques and serial dilution plating, generally require biofilms to be dispersed. Dispersion adds to the well known problems associated with enumeration of planktonic suspensions. An alternative approach for biofilm quantification is the release and measurement of cellular components such as DNA, yet relatively few studies adopt such methods. DNA-based estimates of cell number may be affected by extracellular DNA and will also include the DNA of dead cells. However it may be possible to circumvent these problems by digesting extracellular DNA prior to extraction or by selectively intercalating the DNA of cells with damaged membranes. DNA extracted from biofilms and planktonic cells may be quantified directly or by quantitative PCR that increases sensitivity and enables enumeration of specific cells. We have compared biofilm and planktonic populations in a laboratory simulation of a contaminated aquifer, by quantification of DNA.
Introduction
Biofilms are the predominant mode of growth for bacteria in nature (Costerton and Lewandowski 1995) and are commonly regarded as being more important than planktonic cells with respect to both advantageous and detrimental biological processes in nature and industry. For example, biofilms have often been observed to be more physiologically active in rivers compared to neighbouring planktonic communities, and are thus presumed to play dominant roles in processes such as the biodegradation of pollutants (Araya et al. 2003).
Microbial ecologists seek to understand the interactions between microorganisms and the environment, and in this context biofilms are part of the wider community which includes planktonic cells. In addition engineers are increasingly interested in the role of bacteria in many applications, be they attached or not. In order to satisfy some of the demands of engineers and microbial ecologists, it is therefore necessary for biofilm researchers to be able to estimate cell numbers in both planktonic and biofilm phases.
A major stumbling block for achieving a full understanding of the dynamic interplay between planktonic and sessile populations is a lack of methods for quantifying bacteria, particularly bacteria in biofilms. For laboratory studies microscopy is often the method of choice for quantifying biofilms. However many biofilms are not amenable to this kind of measurement and they are the focus of this article. In particular microscopy is not well suited for thick or irregular biofilms, and is labour intensive making it impractical for many routine monitoring applications.
Partitioning of bacteria between planktonic and biofilm phases
Biofilms are not isolated static communities; they are in dynamic flux with neighbouring planktonic communities and cells in both phases are subject to ecological pressures. Phenotypic responses to changing conditions influence processes of relevance to biofilms such as colonisation, species succession, and dispersal of cells. These events are driven by the physiology of individual cells, and are reflected in observed community structure and function. Understanding the physical conditions and biological processes which govern microbial attachment and detachment are therefore of fundamental importance to integrating the functions of sessile and planktonic cells in dynamic microbial systems such as aquifers.
Microbiologists are increasingly taking interest in the mechanisms that trigger biofilm formation and, more recently, dispersal. For example Sauer et al (2004) showed that a sudden increase in carbon source availability triggers dispersal of Pseudomonas aeruginosa biofilms, and this event is associated with increased expression of flagella. P. aeruginosa cells apparently detach in this case because conditions are favourable and they can avoid competition with their biofilm neighbours by moving into the planktonic phase. Sauer et al (2004) note that biofilm dispersal under favourable conditions has been observed previously, probably because bacteria only benefit nutritionally from attached growth under unfavourable growth conditions for example when nutrients are scarce.
Similarly, Thormann et al (2005) showed that Shewanella oneidensis biofilms rapidly detached when the flow of media was arrested, and they found that the trigger for biofilm dispersal was reduced oxygen tension caused by metabolic activity in the stagnant media.
Investigation of specific cases like these will contribute greatly to our understanding of large-scale microbial ecology and industrial processes, ultimately enabling better prediction of process success and more informed intervention strategies. For example in the bioremediation of contaminated aquifers by natural attenuation, electron donor and electron acceptor concentrations are routinely included in models to calculate the potential for remediation without human intervention. In such cases including consideration of whether bacteria are likely to be attached to the substratum at different spatial and temporal positions in the contaminant plume may enable more accurate predictions to be made. Microbial ecology studies are increasingly including separate community analyses for planktonic and attached communities, reflecting a growing recognition that these communities occupy distinct niches and perform separate ecological functions (e.g. Tresse et al. 2002, Acinas et al. 1997).
We are presently working on the characterisation of microbial communities and their function in contaminated aquifers, particularly with respect to the remediation of these sites by natural attenuation. It is well known that microbial activity is the primary driving force for the natural attenuation of pollution in aquifers, and it is generally believed that most activity can be attributed to the sessile populations (Harvey et al. 1984, Lehman et al. 2001). However groundwater sampled from such sites yields high numbers of bacteria, suggesting that planktonic populations may also have a role to play in natural attenuation. The separate determination of microbial numbers and community structure in microbial systems such as contaminated aquifers may therefore shed light upon the different roles of planktonic and biofilm cells under various conditions, and ultimately such knowledge can be used to inform prediction and management of microbial processes. Presently an increasing number of studies are analysing neighbouring planktonic and biofilm community structures, however few estimate the populations of these communities, perhaps because of a lack of reliable methods. An overview of the strategies that can be employed to quantify both planktonic and biofilm communities is given below.
Quantification of planktonic cells
There are many methods for counting cells in liquid suspensions, and most of these will be familiar to microbiologists. In most cases, quantification of planktonic cells relies upon thorough mixing and separation of cell aggregates before the counting procedure is performed. Once mixed, various methods can be employed for counting cells in suspension, the most familiar being direct microscopy for total cell numbers and dilution-plating methods for cultivable cells. Some of the advantages and disadvantages of different bacterial quantification methods are summarised in Table 1.
Table 1. Comparison of traditional bacterial quantification methods with DNA based methods. Detection limits are approximate and may be altered by method adjustments such as filtering very large volumes for direct cell counts.
| Method | Advantages | Disadvantages | Detection limit (cells / ml) |
| Dilution plating (viable count) | Quick and simple
Detects only viable cells Yields isolates if required Selective counts possible (e.g. selective agars) |
Cultivation dependent
Cell aggregates and biofilms must be dispersed Labour intensive |
100 |
| Direct count (microscopy) | Culture independent
Selective counts possible (various staining methods) |
Samples must be free of debris
Cell aggregates and biofilms must be dispersed |
100 |
| Quantification of total DNA
(e.g. fluorimetry) |
Culture independent
Suitable for cell aggregates and biofilms High throughput |
Interference from extracellular DNA
Poor detection limit |
105 |
| Quantification of target DNA (quantitative PCR) | Culture independent
Sensitive Suitable for cell aggregates and biofilms Selective counts possible (primer selection) High throughput |
Relatively expensive
Primer selection may be difficult Inter-assay variability Contamination problems close to detection limit |
1000 |
Culture-based quantification
Variously called viable cell counts or cultivable cell counts, the process of serially diluting a sample and plating out aliquots onto nutrient-containing agar plates is a useful method for estimating the number of viable cells in planktonic suspensions. Serial dilution methods are based upon the assumption that each viable cell in the sample gives rise to a single colony when it is plated out, but it is well known that this assumption is flawed in many cases. In particular over 99 % of microorganisms appear not be amenable to culture (Pace 1997), and also co-aggregates of cells may not be easily dispersed.
Quantification by optical microscopy
Direct microscopic methods may be performed in a counting chamber or after filtration of the sample, and have the advantage that they are not affected by the cultivability of the sample, and co-aggregated cells can be counted so long as the aggregates are not too large. Another advantage is the possibility of differential staining of different cell types for example by using physiological stains, fluorescent in-situ hybridisation, or antibody based staining methods. Microscopic counting methods are quite time consuming to perform and require the sample to be free from debris.
Alternative methods
Alternatives to microscopic and dilution-plating based counting methods include the use of flow cytometers (e.g. Amann et al.1990) and specialist equipment for measuring respiration or specific cellular constituents such as ATP (e.g. Oulahal-Lagsir et al.2000), or bulk methods such as measurements of turbidity or dry cell weight. In general these methods have to be specifically developed for any given application but once set up they may give reliable and rapid estimates, particularly useful as part of quality assurance strategies for example in food production plants.
Biofilm quantification
One approach to quantify biofilms is to adapt established methods used for planktonic enumeration, with an additional disruption step at the beginning to dislodge the biofilm from the substratum and disperse the cells. Biofilm disruption may be achieved by many methods, including scraping with a scalpel, sonication, and vortex mixing (e.g. Prosser et al.1987; Ferris et al.1989). These adaptations of standard methods can be very useful in many cases, but it must be realised that the limitations are greater than for planktonic quantification because of the extra uncertainty associated with the biofilm disruption step. It is preferable therefore, to avoid methods which rely upon biofilm dispersal in favour of methods which enable direct analysis of the biofilm itself.
Bacterial quantification using biochemical measures
As early as 1973 it was suggested that biochemical components of the cell might be useful in determining biomass from environmental samples (Holm-Hansen 1973). DNA and ATP are of particular interest for this application, however both have potential drawbacks. ATP is not an ideal analog for biomass because its concentration varies depending upon the metabolic activity of the cell, therefore it may be more useful as an indicator of bacterial activity. DNA in many ways is an ideal choice for the estimation of cell numbers since measurement of DNA can essentially be considered a method for counting genomes. However there are potential pit falls with this approach. The advantages and disadvantages of DNA as a measure of biomass are discussed below.
DNA as an estimate of biomass
Measurement of DNA in sediments was suggested as an ideal method for quantification of biomass by Lorenz et al (1981) because the DNA/cell ratio has been found to be quite stable (Kubitschek and Newman 1978; Torsvik and Goksøyr 1978). Earlier work by Holm-Hansen et al (1968) had however found unfeasibly high DNA concentrations in deep water compared to the amount of biomass, and their suggestion that extracellular DNA may be associated with detritus is probably correct. More recently extracellular DNA has been shown to be required for biofilm formation in some cases (Whitchurch et al.2002), and it is also a nutrient source and an intermediate in horizontal gene transfer (Finkel1 and Kolter 2001).
For microbial ecology applications such as community profiling or gene probing, researchers select DNA extraction methods which favour complete representation of the community with minimal shearing of the DNA, often for downstream PCR. These applications do not require quantitative measures of sample DNA, however some workers have recognised the usefulness of such measures. For example Rudney et al (2003) successfully used total biofilm DNA as an estimator for biomass in oral biofilms.
DNA extraction methods for quantification
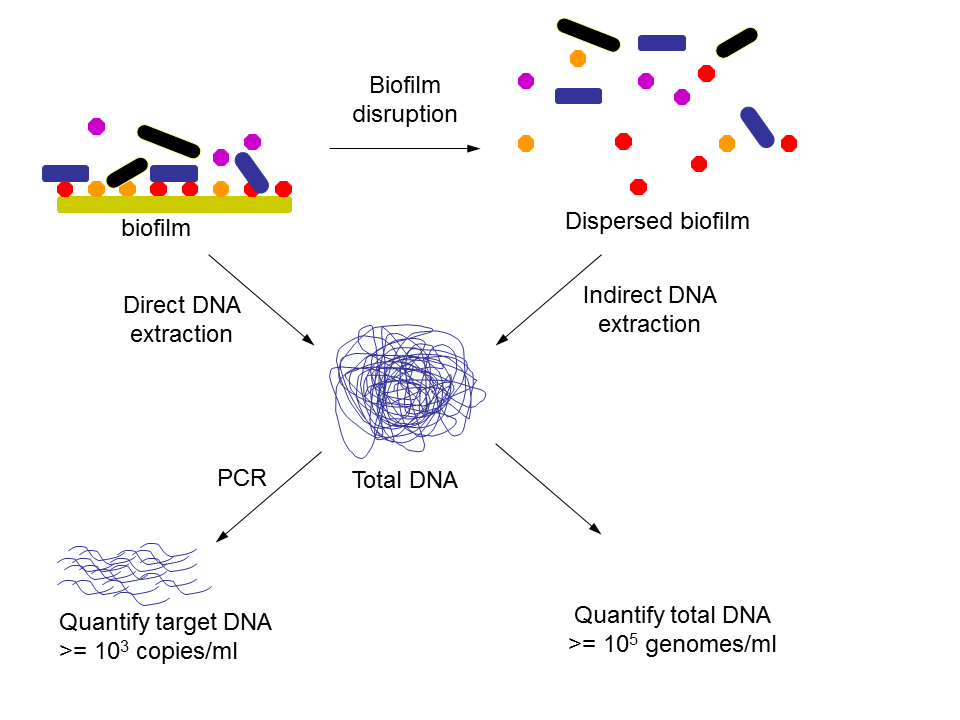
Figure 1. Extraction of DNA from biofilms may be performed directly or after disruption of the biofilm which may be achieved by various means such as scraping and sonication. Extracted DNA may be quantified directly to estimate the number of organisms in a sample, or target DNA may be quantified for all organisms or a subset of organisms using quantitative PCR.
DNA extraction methods may be classified as indirect or direct, depending upon whether cells are extracted from the sample before DNA extraction or not respectively (Figure 1). When DNA is extracted directly from the sample without first separating the cells, DNA yields tend to be higher, but indirect methods give better representation of mixed populations (Gabor et al. 2003).
For the purposes of making biomass estimates direct DNA extraction methods would seem to be more favourable since the loss in yield associated with indirect methods is unlikely to be reproducible, and will impose a higher biomass detection limit.
Direct methods will however co-extract extracellular DNA, so it may be desirable to avoid this by prior treatment with DNAse which has been shown to be effective for water samples (Wegley et al. 2006), however additional development may be required for biofilm samples because sediments may protect DNA from degradation by nucleases (Lorenz et al. 1981). It may also be appropriate to quantify the DNA prior to purification for downstream applications, to avoid possible bias as suggested by Steinberger and Holden (2005).
Most DNA extraction methods will co-extract DNA from both living and dead cells, so interpretation of DNA concentrations for biomass estimation should be considered analogous to total counts rather than viable counts, and this may be problematic for many applications. A potential solution to enable a DNA based alternative to the ‘viable count’ is the selective removal of DNA from dead cells using ethidium monoazide bromide (EMA) treatment, as described by Nocker and Camper (2006). EMA is a DNA intercalating dye which penetrates only cells with compromised cell membrane integrity, whereupon it prevents extraction of the DNA and also renders the DNA resistant to amplification by PCR.
DNA quantification
DNA may be quantified directly using a variety of standard laboratory methods, or specific DNA sequences may be quantified using real-time PCR (Figure 1). Rudney et al (2003) used a picogreen fluorescence assay to determine total DNA and complemented this with a quantitative PCR approach to determine the total number of streptococci in their samples.
The picogreen literature from Invitrogen claims a detection limit of 250 pg ml-1 when used in conjunction with a fluorescence microplate reader, which is equivalent to approximately 105 bacterial genomes per ml. Bacterial quantification by this method therefore has a higher detection limit compared to traditional methods (see Table 1), however a few studies in addition to Rudney et al (2003) have successfully used similar approaches as a means for measuring bacterial biomass. For example Tranvik (1997) found that for lake and sea water, microscopic counts and DNA concentration were correlated, and proposed picogreen fluorimetry as a viable rapid method for assaying bacterial density without even performing a DNA extraction step (i.e. DNA was quantified within whole cells). DNA quantification within cells like this is clearly a rapid and convenient method, however its application is probably limited to clean planktonic samples such as lake water.
We used picogreen fluorescence to determine DNA concentrations from different locations in a laboratory microcosm simulating a phenol contaminated aquifer. Artificial groundwater was pumped through a series of linked sand-filled columns which were inoculated with isolated organisms and arranged to allow sampling of pore water and sand grains with associated biomass from discrete zones (Figure 2). DNA was extracted from pore water (planktonic cells), sand grains (biofilm), and washings from sand grains (loosely attached cells) by lysozyme treatment followed by 40 seconds boiling in STET buffer (8 % sucrose, 5 % triton-X100, 50 mM EDTA, 50 mM Tris.HCl) and vortex mixing. The resulting homogenates were centrifuged to remove large particles of debris, and DNA was quantified from the supernatant by picogreen fluorescence. Serial dilution plating of the liquid samples was also performed.


Figure 2. Linked column microcosm system used to simulate a contaminated aquifer.
Dilution plate counts indicated that there were approximately 107 cells ml-1 in the pore water of the microcosms (planktonic), and washings from the sand grains (loosely attached cells) showed a similar profile but with approximately ten times more cells (data not shown). Relatively high cell numbers easily washed from the sand suggest that many cells may have been immobilised in flocs as an alternative strategy to forming biofilms on the sand.
DNA measurement from pore water and sand washings showed the same pattern of results (Figure 2C), but the amount of DNA detected was higher than predicted from the dilution plating, therefore it seems likely in this case that there was extracellular DNA or dead cells present.
DNA measurement from the sand grains allowed us to estimate the number of cells present in biofilms, which appears to decrease towards the effluent of the microcosm. Calibration of biofilm DNA measurements using data from the pore water and from biofilm washings, indicated an average of approximately 4×106 cells ml-1 attached to sand grains, assuming that the fraction of extracellular DNA remains constant. In conclusion we have shown that quantification of DNA is a useful semi-quantitative measure of microbial biomass. Further development is required to make this truly quantitative.
Quantitative PCR methods have the potential to reduce the detection limit of DNA based bacterial quantification to levels comparable with traditional approaches. For example Panicker et al (2004) reported detection of Vibrio vulnificus at 102 cells ml-1, but Smith et al (2006) in an evaluation of quantitative PCR methods suggest that for this study a detection limit of 103 cells ml-1 may have been more appropriate to discriminate from the no template control (NTC) signal. They go on to recommend inclusion of NTC data when publishing gene copy numbers, and due to inter-assay variation they also point out that care should be taken when comparing data from different experiments.
One limitation of the quantitative PCR approach is the difficulty in selecting suitable target sequences. Total counts can only be accurately achieved using a target gene with conserved copy number for all cell types in the population being assayed, and phylogenetic or functional targets likewise should be selected in the same way. Ribosomal RNA gene copy number is known to vary considerably among bacteria, however to some extent this can be corrected for if the target community composition is known.
Concluding remarks
Table 1 shows a summary of the general advantages and disadvantages associated with traditional bacterial quantification methods, compared with using DNA as a proxy for cell number. The problems with traditional methods are well known, but they are still very useful when used and interpreted with appropriate care. In some cases though, particularly where biofilms and debris are present, the limitations of traditional techniques are compounded and the use of alternative methods for assessing bacterial population size such as DNA quantification should be considered.
References
Acinas, S.G., Rodr|èguez-Valera, F., Pedroès-Alioè, C. (1997) Spatial and temporal variation in marine bacterioplankton diversity as shown by RFLP fingerprinting of PCR amplified 16S rDNA. FEMS Microbiology Ecology 24(1), 27-40.
Amann, R.I., Binder, B.J., Olson, R.J., Chisholm, S.W., Devereux, R., and Stahl, D.A. (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Applied and Environmental Microbiology 56(6), 1919-1925
Araya, R., Tani, K., Takagi, T., Yamaguchi, N., and Nasu, M. (2003) Bacterial activity and community composition in stream water and biofilm from an urban river determined by fluorescent in situ hybridization and DGGE analysis. FEMS Microbiology Ecology 43 (1), 111–119.
Costerton, J.W., Lewandowski, Z., Caldwell, D.E., Korber, D.R., and Lappin-Scott, H.M. (1995) Microbial Biofilms. Annual Review of Microbiology 49, 711-745.
Ferris, F.G., Schultze, S., Witten, T.C., Fyfe, W.S., and Beveridge, T.J. (1989) Metal Interactions with Microbial Biofilms in Acidic and Neutral pH Environments. Applied and Environmental Microbiology 55(5), 1249-1257.
Finkel1, S.E., and Kolter, R. (2001) DNA as a Nutrient: Novel Role for Bacterial Competence Gene Homologs. Journal of Bacteriology 183(21), 6288-6293.
Gabor, E.M., de Vries, E.J., Janssen, D.B. (2003) Efficient recovery of environmental DNA for expression cloning by indirect extraction methods. FEMS Microbiology Ecology 44(2), 153–163.
Harvey, R.W., Smith, R.L., and George, L. (1984) Effect of Organic Contamination upon Microbial Distributions and Heterotrophic Uptake in a Cape Cod, Mass., Aquifer. Applied and Environmental Microbiology 48(6), 1197-1202
Holm-Hansen, O., Sutcliffe, H., and Sharp, J. (1968) Measurement of deoxyribonucleic acid in the ocean and its ecological significance. Limnology and Oceanography 13, 507-514.
Holm-Hansen, O. (1973) The use of ATP determinations in ecological studies. In. Modern methods in the study of microbial ecology bulletin Nr. 17. ed. Rosswall T. pp 215-222. Ecological research committee/NFR, Stockholm.
Kubitschek, H.E., and Newman, C.N. (1978) Chromosome replication during the division cycle in slowly growing, steady-state cultures of three Escherichia coli B/+ strains. Journal of Bacteriology 136, 179-190.
Lehman, R.M., Colwell, F.S., and Bala, G.A. (2001) Attached and Unattached Microbial Communities in a Simulated Basalt Aquifer under Fracture- and Porous-Flow Conditions. Applied and Environmental Microbiology 67(6), 2799–2809.
Lorenz, M.G., Aardema, B.W., and Krumbein, W.E. (1981) Interaction of marine sediment with DNA and DNA availability to nucleases. Marine Biology 64(2), 225-230.
Nocker A., and Camper, A.K. (2006) Selective Removal of DNA from Dead Cells of Mixed Bacterial Communities by Use of Ethidium Monoazide. Applied and Environmental Microbiology 72(3), 1997-2004.
Oulahal-Lagsir, N., Martial-Gros, A., Bonneau, M., Blum, L.J. (2000) Ultrasonic methodology coupled to ATP bioluminescence for the non-invasive detection of fouling in food processing equipment — validation and application to a dairy factory. Journal of Applied Microbiology 89(3), 433–441.
Pace, N.R. (1997) A Molecular View of Microbial Diversity and the Biosphere. Science 276, 734-740.
Panicker, G., Myers, M.L., and Bej, A.K. (2004) Rapid Detection of Vibrio vulnificus in Shellfish and Gulf of Mexico Water by Real-Time PCR. Applied and Environmental Microbiology 70(1), 498-507.
Prosser, B.L., Taylor, D., Dix, B.A., and Cleeland, R. (1987) Method of evaluating effects of antibiotics on bacterial biofilm. Antimicrobial Agents and Chemotherapy 31(10), 1502-1506.
Rudney, J.D., Pan, Y., and Chen, R. (2003) Streptococcal diversity in oral biofilms with respect to salivary function. Archives of Oral Biology 48(7), 475-93.
Sauer, K., Cullen, M.C., Rickard, A.H., Zeef, L.A.H., Davies, D.G., and Gilbert, P. (2004) Characterization of Nutrient-Induced Dispersion in Pseudomonas aeruginosa PAO1 Biofilm. Journal of Bacteriology 186(21), 7312-7326.
Smith, C.J., Nedwell, D.B., Dong, L.F., Osborn, A.M. (2006) Evaluation of quantitative polymerase chain reaction-based approaches for determining gene copy and gene transcript numbers in environmental samples. Environmental Microbiology L.J., 804-815.
Steinberger, R.E., and Holden, P.A. (2005) Extracellular DNA in Single- and Multiple-Species Unsaturated Biofilms. Applied and Environmental Microbiology 71(9), 5404-5410.
Thormann, K.M., Saville, R.M., Shukla, S., and Spormann, A.M. (2005) Induction of Rapid Detachment in Shewanella oneidensis MR-1 Biofilms. Journal of Bacteriology 187(3), 1014-1021.
Torsvik, V.L., and Goksøyr, J. (1978) Determination of bacterial DNA in soil. Soil Biology and Biochemistry 10, 7-12.
Tranvik, L.J. (1997) Rapid Fluorometric Assay of Bacterial Density in Lake Water and Seawater. Limnology and Oceanography, 42(7), 1629-1634.
Tresse, O., Lorrain, M.-J., Rho, D. (2002) Population dynamics of free-floating and attached bacteria in a styrene-degrading biotrickling filter analyzed by denaturing gradient gel electrophoresis. Applied Microbiology and Biotechnology 59(4-5), 585-590.
Wegley, L., Mosier-Boss, P., Lieberman, S., Andrews, J., Graff-Baker, A., and Rohwer, F. (2006) Rapid estimation of microbial numbers in water using bulk fluorescence. Environmental Microbiology 8(10), 1775–1782.
Whitchurch, C.B., Tolker-Nielsen, T., Ragas, P.C., Mattick, J.S. (2002) Extracellular DNA Required for Bacterial Biofilm Formation. Science 295, 1487.